RESUMEN
ANTECEDENTES: La asociación MURCS en una afección rara que comprende alteraciones de los conductos de Müller, aplasia renal y malformaciones esqueléticas, sobre todo cervicotorácicas. La mayor parte se diagnostica en la adolescencia por amenorrea primaria y el tratamiento requiere de la participación de varios especialistas.
CASO CLÍNICO: Adolescente de 14 años, con antecedente de riñón en herradura diagnosticado en la etapa prenatal. Al nacimiento, se apreciaron: paladar ojival, epicanto interno y puente nasal amplio. El cariotipo reportado fue 46XX. Acudió a consulta debido a la falta de menstruaciones. Exploración física: talla baja, cuello corto, con escoliosis. Mamas y vello púbico en Tanner III. El ultrasonido reportó: ectopia renal cruzada y ausencia de útero. Con la resonancia magnética se confirmó la agenesia útero-vaginal y la ectopia renal cruzada.
CONCLUSIÓN: La asociación MURCS es una enfermedad congénita rara, con una gran diversidad de manifestaciones clínicas. El diagnóstico puede sospecharse en etapas muy tempranas, como en el caso expuesto. El ultrasonido y la resonancia magnética tienen alta sensibilidad diagnóstica. El tratamiento debe ser individualizado y multidisciplinario, enfocado en aliviar los síntomas ginecológicos y urológicos, favorecer la función sexual, brindar alternativas de fertilidad y apoyo psicológico.
PALABRAS CLAVE: Asociación MURCS, síndrome de Mayer-Rokitansky-Küster-Hauser tipo II, aplasia o disgenesia mülleriana
ABSTRACT
BACKGROUND: The MURCS association is a rare pathology that includes alterations of the Müllerian ducts, renal aplasia and skeletal malformations, especially cervicothoracic. Most are diagnosed in adolescence due to primary amenorrhea and treatment involves several specialists.
CLINICAL CASE: 14-year-old adolescent with a history of horseshoe kidney diagnosed prenatally. At birth, a high-arched palate, internal epicanthus and broad nasal bridge were noted; a karyotype was requested, which reported 46XX. She came for primary amenorrhea. Physical examination: short stature, short neck with scoliosis. Breasts and pubic hair in Tanner III. Ultrasound reports crossed fused renal ectopia and absence of uterus. With magnetic resonance imaging, uterovaginal agenesis and crossed renal ectopia were confirmed.
CONCLUSION: MURCS association is a rare congenital disease, with a great diversity of clinical manifestations. Diagnosis can be suspected at early stages, as in the present case. Ultrasound and magnetic resonance imaging have high diagnostic sensitivity. Treatment should be individualized and multidisciplinary, focused on alleviating gynecological and urological symptoms, promoting sexual function, providing fertility alternatives and psychological support.
KEYWORDS: MURCS Association, Mayer-Rokitansky-Küster-Hauser syndrome type II, Mullerian Aplasia-Dysgenesis
ANTECEDENTES
Las alteraciones derivadas de los conductos müllerianos incluyen diversas malformaciones que pueden ser menores o incluir agenesia de útero y vagina. Una de las más conocidas es el síndrome de Mayer-Rokitansky-Küster-Hauser que se caracteriza por ausencia de útero y las dos terceras partes superiores de la vagina. La incidencia de este síndrome aislado (tipo I) se estima en 1 caso por 4000 nacidas vivas,1 pero también puede estar asociado con otras malformaciones (MRKH tipo II), con una incidencia de 1 caso por cada 50000 mujeres.2
A partir de 1948 a la fecha se han descrito alteraciones asociadas con la agenesia útero-vaginal, sobre todo anomalías renales que se encuentran en 25 al 50% de todos los casos de Mayer-Rokitansky-Küster-Hauser.3 En 1976 Griffin describió que existían, al menos, 17 series reportadas, por lo que se habían acumulado 560 casos, de los que el 34% tenían alteraciones renales y el 12% alteraciones esqueléticas.4 En 1979 Duncan y colaboradores reportaron 30 casos con esta asociación y adoptaron el acrónimo MURCS, por sus siglas en inglés para aplasia del conducto de Müller (MU), aplasia renal (R) y displasia de las somitas cervicotorácicas (CS). Concluyeron que todas tenían talla baja (menor a 153 cm), con aplasia o hipoplasia del conducto de Müller (96%), agenesia o ectopia renal (80%) y anomalías cervicotorácicas (80%).1
Su causa se desconoce, quizá sea heterogénea, con varias hipótesis propuestas sin evidencia suficiente para establecer una verdadera causa. La mayoría de los casos reportados han sido esporádicos; sin embargo, existen algunos con recurrencia familiar.1,5 La diversidad del cuadro clínico hace suponer que existen casos que no se reportan debido a que los defectos son menores. Se han propuesto mutaciones del gen WNT4, pero no hay suficientes casos que sustenten esta teoría.6 Los reportes más recientes proponen anomalías cromosómicas, como las duplicaciones intersticiales o las deleciones, aún sin evidencia contundente.7,8
Si bien la causa no es precisa, la alteración primaria ocurre en la cuarta semana de la embriogénesis, periodo en el que los nefrotomos y las somitas cervicotorácicas del mesodermo paraxial tienen una relación estrecha.
El diagnóstico puede sospecharse tempranamente, incluso desde la etapa prenatal si hay alteraciones renales o esqueléticas; sin embargo, suele establecerse en la adolescencia, pues el 85% consultará por amenorrea primaria.9
Del 2 al 30% de las mujeres pueden padecer dismenorrea, distensión abdominopélvica o dolor con amenorrea, cuando hay rudimentos uterinos con estenosis cervical o falta de unión con la vagina; incluso algunas de estas mujeres pueden resultar con leiomiomatosis y adenomiosis.10
La atención médica debe ser multidisciplinaria e individualizada y es de suma importancia conocer las recomendaciones que deben darse a las pacientes y a sus familias. La mayor parte de los artículos abordan de manera efímera el tratamiento, es por eso que el objetivo central de este trabajo es reportar el caso de una paciente con asociación MURCS, en virtud de que hay pocos casos comunicados en México y exponer las directrices para el tratamiento integral.
CASO CLÍNICO
Adolescente de 14 años, que acudió a consulta debido a la amenorrea primaria. Antecedentes relevantes: embarazo conseguido mediante técnicas de fertilización in vitro. Durante la etapa prenatal se le diagnosticó riñón en herradura. En los primeros días de nacida el ultrasonido reportó: ectasia pielocalicial y requirió cateterismo. En la exploración neonatal se apreciaron fisuras palpebrales antimongoloides, paladar ojival, epicanto interno, puente nasal amplio y genitales externos femeninos. El cariotipo 46XX, sin alteraciones estructurales. La valoración audiológica fue normal. A los 8 y 10 años requirió cateterismo ureteral por síntomas urológicos. La radiografía de columna cervicotorácica por escoliosis, tomada a los 12 años, reportó una fusión costal posterior derecha del primero al cuarto arco, hemivértebras C6, C7 y T1, además de fusión de los cuerpos vertebrales de C2 y C3. Ausencia de apófisis espinosas de C5 hasta C7. Figuras 1 y 2
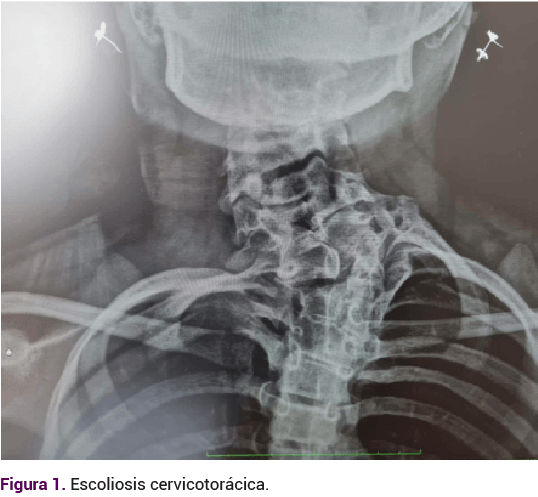
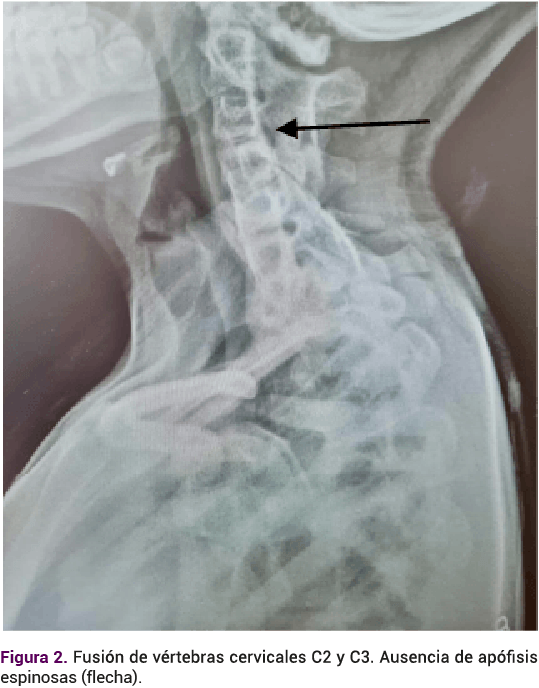
Exploración física al momento de la consulta: fenotipo femenino, sin rasgos faciales anormales, talla 1.46 m, delgada, cuello corto y con ligera desviación derecha de la columna cervical. Desarrollo puberal de mamas y vello púbico en Tanner III. No obstante encontrarse en su rango de edad para esperar la menarquia, se decidió indicar los estudios para confirmar la asociación MURCS debido a los antecedentes de alteraciones renales y cervicales.
El ultrasonido renal reportó ectopia renal cruzada, de apariencia fusionada con el uréter distal único. El ultrasonido pélvico reportó la ausencia del útero y de los ovarios. El perfil hormonal se informó con: FSH 4.39 mUI/mL, LH 2.89 mUI/mL, estradiol 80.5 pg/mL, androstenediona 1.27 ng/mL, testosterona total 0.2 pg/mL, prolactina 14.2 ng/mL, TSH 1.1 mUI/mL. La resonancia magnética reportó: ausencia de útero, ambos ovarios con desplazamiento cefálico, sin evidencia de imagen que sugiriera el canal vaginal (Figura 3). Riñón único intrapélvico parasagital izquierdo (Figura 4) con diagnóstico por imagen tomográfica de ectopia renal cruzada y agenesia úterovaginal.
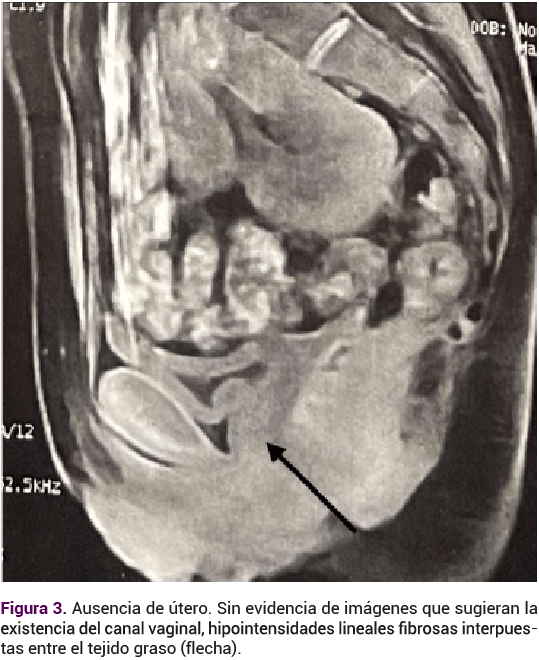
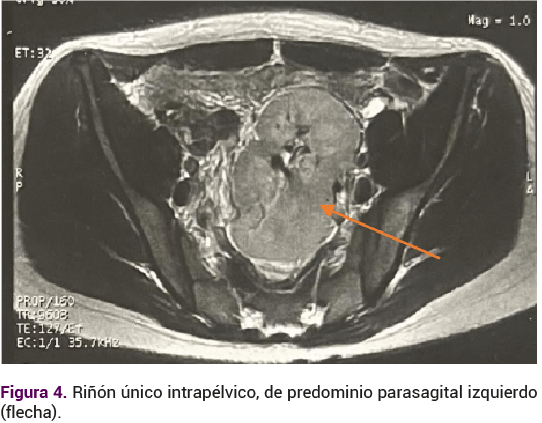
Se integró el diagnóstico de asociación MURCS, se explicó la afección a los padres y se expusieron las recomendaciones actuales para la atención integral; sin embargo, decidieron posponer todo tipo de tratamiento ginecológico y psicológico hasta que la paciente cumpliera la mayoría de edad. En la actualidad permanece en vigilancia anual del urólogo. Los padres aceptaron compartir el material y aceptaron que se publicara el caso.
DISCUSIÓN
La asociación MURCS es una enfermedad congénita rara. En 1993 Strübbe y colaboradores hicieron una revisión de 91 pacientes con síndrome de Mayer-Rokitansky-Küster-Hauser y encontraron 34 pacientes con anomalías renales.11 Griffin y Mahajan reportaron en sus series un porcentaje de alteraciones renales parecido, 43 y 28%, respectivamente.4,12 En 2006 Oppelt y su grupo conjuntaron 18 series de pacientes con Mayer-Rokitansky-Küster-Hauser con 521 casos, 32% con anomalías renales y 12% con asociación MURCS.5 Un estudio multicéntrico más reciente del 2021 con 1055 pacientes reportó que el 69.6% tenían síndrome de Mayer-Rokitansky-Küster-Hauser tipo I y el 30.4% tipo II.13 Por lo anterior se sugiere que todas las pacientes con agenesia útero-vaginal deben ser estudiadas para descartar otras alteraciones, sobre todo renales. Pero si se diagnostica primero alguna malformación renal asociada con alteraciones esqueléticas es necesario investigar la integridad del útero y la vagina; como sucedió en la paciente del caso.
El diagnóstico puede sospecharse en etapas muy tempranas, desde el periodo fetal, neonatal o en la infancia. En la paciente del caso se diagnosticó riñón en herradura prenatalmente, pero se consideró malformación única y el diagnóstico definitivo se estableció en la adolescencia. Al igual que la mayoría de las pacientes consultó por amenorrea primaria.
Los casos clínicos reportados más recientemente describen un cuadro clínico variado, con amenorrea primaria hasta el 85%, alteraciones renales 18%, malformaciones esqueléticas 13%, dolor pélvico 4%, infertilidad 2% y en muy pocos casos el diagnóstico se estableció durante la necropsia.14-20 La edad promedio al diagnóstico es de 16 años, en la paciente del caso fue a los 14 años.
Los tres criterios diagnósticos para integrar esta asociación son:
Aplasia congénita del útero y la parte superior (2/3) de la vagina en mujeres con desarrollo normal de caracteres sexuales secundarios y un cariotipo normal de 46, XX. La agenesia vaginal completa ocurre en el 75% de los casos mientras que el 25% restante tiene una vagina reducida.10
Alteraciones renales: agenesia unilateral, ectopia o riñón en herradura. El riñón ectópico es la alteración más frecuente 40-50% (con riñón contralateral normal 30%, ausente 12%). Agenesia renal 43%. Anomalías de la pelvis renal o uréteres 12%. Atrofia renal 5%.4,21,22
Alteraciones esqueléticas. La afección más común es la escoliosis en la columna cervical (17%), pero suelen ser diversas, como agenesia de toda la vértebra, hemivértebras segmentadas, fusión vertebral o vértebra en bloque (anomalía de Klippel-Feil) 4%.4,23,24
La paciente del caso cumplió con los tres criterios: ausencia de útero y vagina, ectopia renal, escoliosis y malformaciones vertebrales, además, talla baja.
Los defectos auditivos suelen manifestarse en el 10 al 25% de los casos25 y otras malformaciones menos comunes, como el ano imperforado o la atresia anal.26
El ultrasonido pélvico transabdominal es el estudio de primera elección para evaluar a las pacientes con sospecha de alteración mülleriana. Sin embargo, la resonancia magnética es el estudio más sensible y específico. Debe practicarse cuando los hallazgos ecosonográficos no sean concluyentes o, bien, para confirmar el diagnóstico. Además, tiene la ventaja de reconocer si existen rudimentos uterinos que no pudieron identificarse en el ultrasonido. Permite una evaluación precisa de la aplasia uterina y se pueden identificar malformaciones renales y esqueléticas asociadas.25 Otros estudios, como la cistoscopia, la vaginoscopia y la laparoscopia están reservados para evaluar la anatomía cuando existan anomalías complejas y difíciles de diagnosticar.
Los estudios de laboratorio, como los perfiles hormonales y el cariotipo, son de utilidad para excluir diagnósticos diferenciales, sobre todo en la agenesia uterina. El diagnóstico diferencial debe establecerse en las pacientes con amenorrea primaria, como en el caso de atresia vaginal aislada, síndrome de Turner, síndrome de WNT4 o síndrome de insensibilidad a los andrógenos.25 En la paciente del caso, el estudio hormonal reportó valores normales.
El tratamiento de las pacientes con asociación MURCS debe ser individualizado y multidisciplinario, puede requerir la participación de varios especialistas: pediatra, genetista, radiólogo, ginecólogo, urólogo, ortopedista, neurólogo y psicólogo. Debe estar enfocado en cuatro aspectos fundamentales: aliviar los síntomas ginecológicos o urológicos, favorecer la función sexual, exponer las alternativas de fertilidad y, lo más importante, brindar el apoyo psicológico.
Es importante inspeccionar los genitales externos con el consentimiento de la paciente y, en su caso, de los padres para evaluar el clítoris, los labios mayores y menores, sobre todo el introito, que puede tener un rodete himeneal normal y una vagina de longitud variable, pero a menudo menor de 2 cm; o bien, sin rodete himeneal cuando la aplasia vaginal es completa.25 La longitud vaginal puede evaluarse de manera digital, o bien con sonda urinaria, o dilatador de Hegar.
Una paciente con útero hipoplásico, con cuello uterino estenótico o pobre unión con la vagina, suele requerir la vaginoplastia, drenaje uterino y la dilatación cervical en el momento que haya dolor pélvico o distensión abdominal.27
Los beneficios y riesgos potenciales de las intervenciones quirúrgicas deben evaluarse con las pacientes y, en la mayoría de los casos, con sus padres, teniendo un enfoque de respecto a sus deseos, creencias y tradiciones culturales. Debe respetarse el tiempo suficiente entre el diagnóstico y la decisión del tratamiento de la aplasia vaginal.
En el momento en que la paciente exprese interés en su vida sexual deben informársele las alternativas disponibles, explicar los métodos quirúrgicos y no quirúrgicos de alargamiento vaginal. No se recomienda este tratamiento en niñas, pues primero deben comprender su condición y saber que el procedimiento puede ser largo, complejo y doloroso.
El método no quirúrgico para crear una neovagina consiste en: dilatación instrumental, que debe proponerse de primera intención, no causa cicatrices y conserva una lubricación vaginal normal. Si el cumplimiento es adecuado, los resultados funcionales son buenos en menos de seis meses porque más del 85% de los casos refieren relaciones sexuales satisfactorias y dependerá de la motivación y constancia de cada paciente. Puede ser un proceso que lleve un largo tiempo, desde dos meses hasta más de dos años.25 También está condicionado a la longitud vaginal inicial, que puede ser de 0.5 a 7 cm.27
En caso de fracaso o rechazo del método de dilatación instrumental puede recurrirse a diferentes técnicas quirúrgicas que, generalmente, requieren dilataciones posoperatorias. La principal limitación es que muy pocos cirujanos practican este tipo de procedimientos.
En los casos de deseo reproductivo es importante orientar a las pacientes explicándoles las técnicas de reproducción asistida, con gestación subrogada o, bien, la adopción o el trasplante de útero, cuya técnica ha tenido casos exitosos en Europa desde el 2014.25
El tratamiento de las malformaciones vertebrales está enfocado en el control del dolor y medidas conservadoras. Se han reportado casos de hipoperfusión o eventos trombóticos que pueden ocasionar infartos. El tratamiento quirúrgico se justifica en casos específicos cuando el movimiento cervical está asociado con lesiones de la médula espinal o con infartos por hipoperfusión.28
Cuando las alteraciones renales ocasionan un deterioro de la función es necesaria la consulta con el urólogo para una evaluación detallada. En algunos casos pueden ser necesarias múltiples intervenciones, incluso, la nefrectomía. El pronóstico dependerá de la función renal y de las alteraciones de cada paciente.
Como parte de la atención médica integral es necesaria una evaluación psicológica porque el diagnóstico de esta enfermedad puede ser devastador para algunas pacientes, desde el momento en que se concientizan que no van a tener nunca una menstruación ni concepción natural, eso puede causarles un trauma, ansiedad y, en ocasiones, baja autoestima. En la actualidad existen grupos de apoyo internacionales, como AMAR (Asociación de Apoyo a Mujeres con Aceptación del síndrome de Rokitansky) en España, o MAYNA, en Argentina, en donde varias mujeres pueden compartir su historia para ayudar a otras con este diagnóstico, incluso en forma virtual.
CONCLUSIONES
La asociación MURCS es una enfermedad rara, con una gran diversidad de manifestaciones clínicas. No existe aún una evidencia que demuestre su causa. Identificar las alteraciones involucradas es fundamental para una adecuada atención médica. El diagnóstico tiene una gran repercusión psicológica en la paciente y su familia. Conocer las recomendaciones para el tratamiento permite dar una información completa y clara para el futuro de las mujeres con esta enfermedad.
REFERENCIAS
- Duncan PA, Shapiro LR, Stangel JJ, Klein MR, et al. The MURCS association: müllerian duct aplasia, renal aplasia, and cervicothoracic somite dysplasia. J Pediatr 1979; 95: 399-402. https://doi.org/10.1016/s0022-3476(79)80514-4
- Witchel SF. Disorders of sex development. Best Pract Res Clin Obstet Gynaecol 2018; 48: 90-102. https://doi.org/10.1016/j.bpobgyn.2017.11.005
- Pinsky L. A Community of Human Malformation Syndromes Involving the Müllerian Ducts, Distal Extremities Urinary Tract and Ears. Teratology 1973; 9: 65-80. https://doi.org/10.1002/tera.1420090109
- Griffin JE, Edwards C, Madden JD, et al. Congenital absence of the vagina. Ann Intern Med 1976; 85 (2): 224-36. https://doi.org/10.7326/0003-4819-85-2-224
- Oppelt P, Renner SP, Kellermann A, Brucker S, et al. Clinical aspects of Mayer-Rokitansky-Küster-Hauser Syndrome: Recommendations for clinical diagnosis and staging. Hum Reprod 2006; 21 (3): 792-97. https://doi.org/10.1093/humrep/dei381
- Shoar Z, Ganguly T, Anderson C, et al. Absence of WNT4 gene mutation in a patient with MURCS association. JPEM 2014; 27 (5-6): 555-559. https://doi.org/10.1515/jpem-2013-0335
- Hofstetter G, Concin N, Marth C, et al. Genetic analyses in a variant of Mayer-Rokitansky-Kuster-Hauser syndrome (MURCS association). Wien Klin Wochenschr 2008; 120: 435-39. https://doi.org/10.1007/s00508-008-0995-4
- Acién P, Galán F, Manchón I, et al. Hereditary renal adysplasia, pulmonary hypoplasia and Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: a case report. Orphanet J Rare Dis 2010; 5: 1-6. https://doi.org/10.1186/1750-1172-5-6
- Centre de Référence des Pathologies Gynécologiques Rares. Synthèse à destination du médecin traitant. Syndrome de Mayer-Rokitansky-Kuster-Hauser. 2021. https://www.has-sante.fr/upload/docs/application/pdf/2021-11/synthese_mg_syndrome_de_mayer_rokitansky_kuster_hauser.pdf
- Urbina RN, Serrano EN, Solís SM. Interpretation of Mayer-Rokintansky-Küster-Hauser syndrome by systematic literature review. Salud, Ciencia y Tecnología 2024; 4: 930. https://doi.org/10.56294/saludcyt2024930
- Strübbe EH, Willemsen WN, Lemmens JA, et al. Mayer-Rokitansky-Küster-Hauser syndrome: distinction between two forms based on excretory urographic, sonographic, and laparoscopic findings. AJR 1993 ;160: 331-4. https://doi.org/10.2214/ajr.160.2.8424345
- Mahajan P, Kher A, Khungar A, et al. MURCS association. A review of 7 cases. J Postgrad Med 1992; 38 (3): 109-111. PMID: 1303407
- Chen N, Pan H, Luo G, et al. Clinical characteristics of 1,055 chinese patients with Mayer-Rokitansky-Küster-Hauser syndrome: a nationwide multicentric study. Fertility and Sterility 2021; 116 (2): 558-65. https://doi.org/10.1016/j.fertnstert.2021.02.033
- Ostia GP, Jiménez DL, Plaza BL. Asociación MURCS: reporte de caso de un recién nacido de sexo femenino. Perinatol Reprod Hum 2022; 36: 2. https://doi.org/10.24875/per.21000001
- Lee KS, Kim DS, et al. Long-term follow-up on MURCS (Müllerian duct, renal, cervical somite dysplasia) association and a review of the literature. Ann Pediatr Endocrinol Metab 2019; 24: 1-5. https://doi.org/10.6065/apem.2019.24.3.1
- Kumar S, Sharma S. MURCS (Müllerian duct aplasia-renal agenesis-cervicothoracic somite dysplasia): a rare cause of primary amenorrhoea. Oxf Med Case Reports 2016; 4: 73-5. https://doi.org/10.1093/omcr/omw022
- Sharmila V, Kalluri S, Yoga P, et al. Mayer-Rokitansky-Kuster-Hauser Syndrome type II with Mullerian duct dplasia-Renal Dysplasia-Cervical somite anomalies association: A case report and review of literature. Bangladesh Journal of Endocrinology and Metabolism 2024; 3(2):61-64. https://doi.org/10.4103/bjem.bjem_4_24
- Jahan Shirin, Akter S. Partial mullerian agenesis with renal and skeletal anomalies: an observational study of MURCS association. Sch Int J Obstet Gyne 2022; 5 (1): 1-6. https://doi.org/10.36348/sijog.2022.v05i01.001
- Bellal AR, Shirbur P, Geetha RG. Mayer-Rokitansky-Kuster-Hauser Syndrome - A detailed study of nine cases. J Evid Based Med Healthc 2020; 7: 2479-84. https://doi.org/10.18410/jebmh/2020/513
- Cobb A, Fisher CL. Cadaveric case report of Mayer-Rokitansky-Küster-Hause (MRKH) syndrome type II. Transl Res Anatomy 2024; 36: 100310. https://doi.org/10.1016/j.tria.2024.100310
- Soekersi H, Trihadrian R. Mayer-Rokitansky-Kuster-Hauser syndrome type II with crossed fused renal ectopia: A rare case report. Radiology Case Reports 2023; 18 (5): 1877-81. https://doi.org/10.1016/j.radcr.2023.01.107
- Wu CQ, Childress KJ, Traore EJ, et al. A review on müllerian anomalies and their Urologic association. Pediatric Urology 2021; 151: 98-106. https://doi.org/10.1016/j.urology.2020.04.088
- Katam KK, Satapathy D, Arumulla M. M MURCS syndrome: atypical form of Mayer-Rokitansky-Kuster-Hauser Syndrome. J Obstet Gynecol India 2024. https://doi.org/10.1007/s13224-023-01916-y
- Álvarez UM, Sáiz A, Álvarez S, et al. Displasias vertebrales múltiples asociadas a síndrome de Rokitansky: una causa poco frecuente de escoliosis congénita. A propósito de un caso. Rehabilitación (Madr) 2010; 44 (3): 261-66. https://doi.org/10.1016/j.rh.2010.02.003
- Morcel K, Camborieux L. Programme de Recherches sur les Aplasies Müllériennes (PRAM). Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome-Orphanet J Rare Dis 2007; 2: 13. https://doi.org/10.1186/1750-1172-2-13
- Morao S, Chaves F, Virella D, et al. Asociación MURCS y malformación anorrectal: Informe de caso de una recién nacida. J Pediatr Surg Case Rep 2017; 18: 19-23. http://dx.doi.org/10.1016/j.epsc.2017.01.004
- Carranza-Lira S, Forbin K, Martínez-Chéquer JC. Rokitansky syndrome and MURCS association--clinical features and basis for diagnosis. Int J Fertil Womens Med 1999; 44 (5): 250-5. PMID: 10569454
- Santos M, Cruz S, Casimirio C, et al. Disección de la arteria vertebral asociada a síndrome MURCS. Rev Neurol 2017; 64 (4):190-92. https://doi.org/10.33588/rn.6404.2016336


