RESUMEN
ANTECEDENTES: La trasposición de las grandes arterias es un padecimiento común, con una alta tasa de morbilidad y mortalidad atribuidas, el diagnóstico prenatal temprano es importante para mejorar la supervivencia posnatal mediante la corrección quirúrgica.
CASO CLÍNICO: Paciente de 22 años, con reporte ecográfico de detalle anatómico: transposición de grandes vasos y restricción del crecimiento intrauterino. Estudios genéticos con cariotipo normal y microarray con microdeleción de 1.4Mb del cromosoma 1q21.1. Nació con dismorfia facial asociada a cardiopatía.
CONCLUSIÓN: La microdeleción 1q21.1 es una alteración cromosómica rara, en la que se ven afectados múltiples genes, anomalía que se traduce en una amplia variedad de alteraciones fenotípicas, lo que denota la importancia de los estudios genéticos como método de elección para su identificación, además de poder resaltar la importancia de la medicina 5P.
PALABRAS CLAVE: Cardiopatías congénitas mayores, transposición de las grandes arterias, diagnóstico prenatal temprano, microdeleción, genómica, bioinformátic
ABSTRACT
BACKGROUND: Transposition of the great arteries is a common condition associated with high morbidity and mortality. Early prenatal diagnosis is important to improve postnatal survival by surgical correction.
CLINICAL CASE: 22-year-old female patient with ultrasound findings of transposition of the great arteries and intrauterine growth restriction. Genetic studies with normal karyotype and microarray with microdeletion of 1.4Mb of chromosome 1q21.1. He was born with facial dysmorphia associated with heart disease.
CONCLUSION: 1q21.1 microdeletion is a rare chromosomal alteration involving multiple genes, an anomaly that leads to a wide variety of phenotypic alterations, highlighting the importance of genetic studies as the method of choice for its identification, in addition to highlighting the importance of 5P medicine.
KEYWORDS: Major congenital heart disease, Transposition of the great arterie, Early prenatal diagnosis, Microdeletion, Genomics, Bioinformatics
ANTECEDENTES
La trasposición de las grandes arterias es una cardiopatía congénita importante, caracterizada por una discordancia ventrículo-arterial en el contexto de conexiones auriculoventriculares normales.1 Es una afección en la que la aorta sale del ventrículo derecho y la arteria pulmonar proviene del ventrículo izquierdo.2 Representa del 5 al 7% de todas las cardiopatías congénitas; es la más común de las enfermedades cianóticas, con una incidencia de 2 a 3 casos por cada 10,000 nacidos vivos y se asocia, pobremente, con anomalías extracardiacas. Es más frecuente en varones con una relación 2:1 y su tasa de diagnóstico prenatal entre 40 al 80% debido a la visualización ecocardiográfica de los conductos de salida.3
Se han descrito dos tipos de trasposición de las grandes arterias: una forma simple con discordancia ventriculoarterial, sin otras anomalías cardiacas y la forma compleja con asociación de defectos septales ventriculares (45% asociados a CIV, 15% sin alteración hemodinámica), obstrucción de la vía de salida ventricular 20-30%, coartación de la aorta 5%. La supervivencia se estima entre 75 a 80% o 98 a 99%, dependiendo del tipo de anomalía en las arterias coronarias.1
Puesto que la trasposición de las grandes arterias es un padecimiento común, con una alta tasa de morbilidad y mortalidad atribuidas, el diagnóstico prenatal temprano es importante para mejorar la supervivencia posnatal mediante la corrección quirúrgica. Su causa exacta sigue sin conocerse por completo y rara vez se asocia con aberraciones cromosómicas numéricas y anomalías extracardiacas.4 Entre las microdeleciones o duplicaciones estudiadas para esta anomalía, se han descrito en diferentes regiones: 22q11, 16p11.2 y 1q21.1. Las microdeleciones en 1q21.1 rara vez se encuentran en la población general y se ha descrito una expresividad clínica variable, incluidas las alteraciones en el desarrollo motor, trastornos psiquiátricos, microcefalia, alteraciones cardiacas y en la talla, convulsiones y alteraciones renales.5
Hasta la fecha, la investigación genética se ha basado en el estudio de pacientes con una misma enfermedad o características clínicas similares e investigación de los cambios genéticos que podrían ser los responsables. Esto ha permitido, a lo largo de la historia, identificar las causas genéticas de múltiples enfermedades y los mecanismos moleculares implicados.6 Este concepto, llamado fenotipado inverso, tiene un especial interés en la medicina preventiva, con el fin de poder ofrecer a las pacientes un consejo previo a la concepción y, finalmente, el mejor tratamiento según el padecimiento en estudio.6
El caso aquí reportado describe una cardiopatía congénita mayor, asociada con una micropérdida en el cromosoma 1q21.1, como un caso de interés en la correlación genotipo-fenotipo que busca resaltar la importancia de la medicina 5P en la que se busca una asesoría personalizada, según el diagnóstico del paciente, predictiva, preventiva, a través de la consejería previa a la concepción, participativa y poblacional, en donde se den alternativas de tratamiento, seguimiento y pronóstico.7
CASO CLÍNICO
Paciente primigesta de 22 años, sin antecedentes familiares de importancia. Durante el seguimiento del control prenatal, a las 22.2 semanas de embarazo, en la ecografía de detalle anatómico se advirtió un hallazgo anormal. Se reportó un feto en crecimiento, en percentil 11, con peso de 424 g, hueso nasal hipoplásico y cardiopatía compleja (transposición de grandes arterias), además de comunicación interventricular perimembranosa. Durante el seguimiento ecográfico del bienestar fetal, en las ecografías de las semanas 23 y 26, se advirtió la restricción del crecimiento intrauterino (percentil 2) con Doppler normal, además de la cardiopatía ya mencionada en la ecografía y ecocardiografía.
Debido a los hallazgos se practicó una amniocentesis diagnóstica, cariotipo en bandas G e hibridación genómica comparativa. La primera con reporte normal 46XY y la segunda con reporte anormal arr [GRCh38] 1q21.1q21.2 (147,029,795_148,437,975) X1 interpretado como una microdeleción en la región cromosómica 1q21.1.
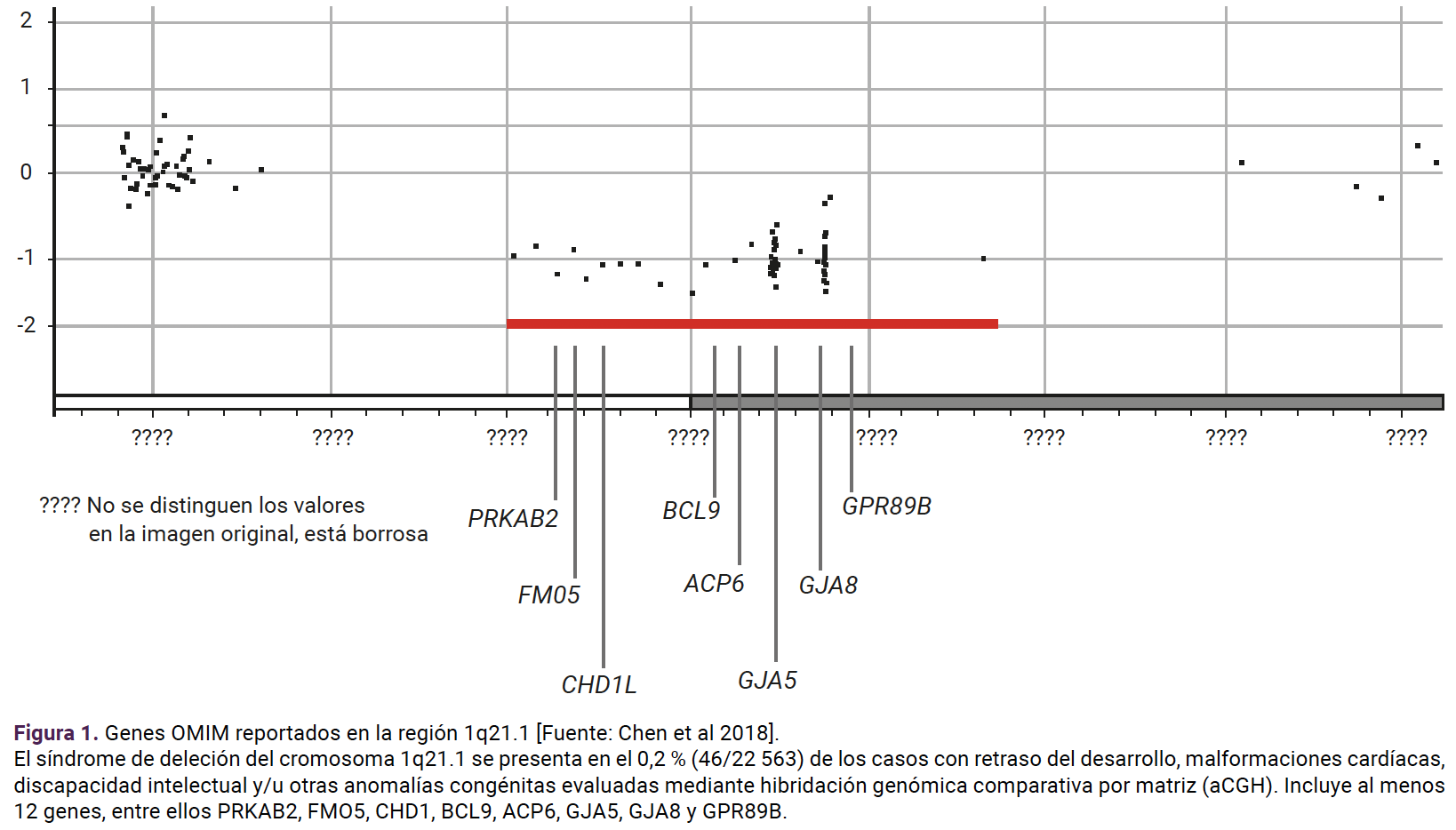
Matriz de puentes genómicos comparativos aCGH

Se identificó una microdeleción en la región cromosómica 1q21.1 de 1,4 Mb que se clasificó patógena con afectación a 36 genes HGNC y 8 genes de referencia OMIM. Se trata de una microdeleción recurrente reportada en la bibliografía.
Análisis de interacción génica
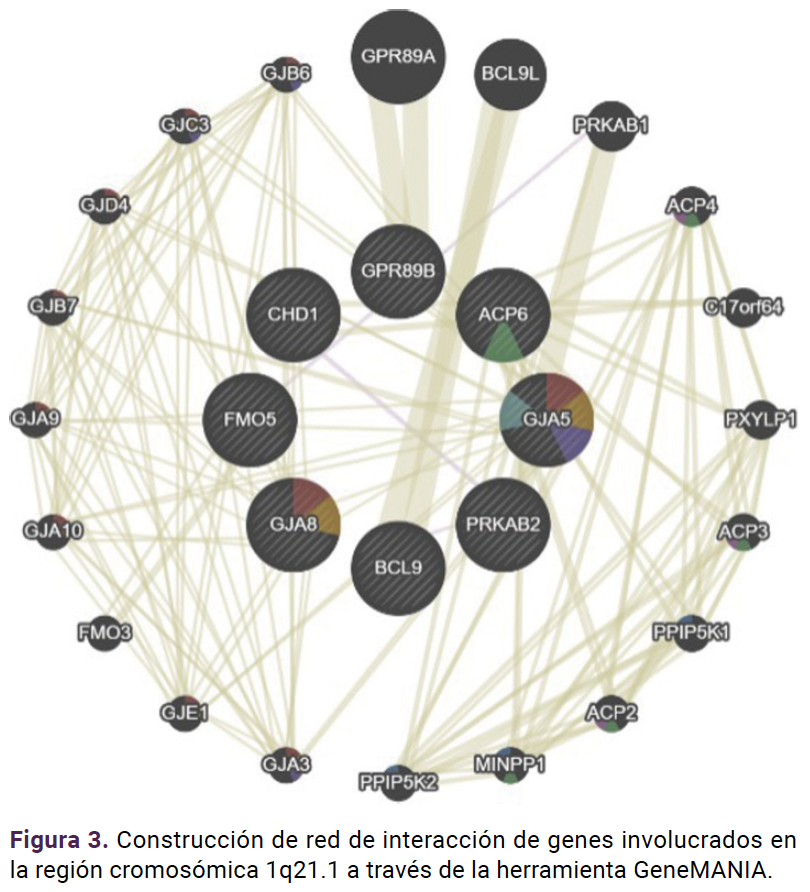
Con uso de la base de datos del Sistema de Predicción de Interacciones Funcionales de Genes, obtenida GeneMANIA, se evaluaron las redes de expresión de las interacciones físicas, co-expresión, predicción, rutas, co-localización, interacciones genéticas y dominios proteicos de los genes. Si bien estos genes, en conjunto, no presentan interacción alguna, individualmente se relacionan con uniones en hendidura, unión célula-célula, actividad de fosfatasas, organización lítica vacuolar y diversos procesos metabólicos de interés. Figura 2

Luego de haber confirmado el diagnóstico se otorgó el asesoramiento genético respectivo y la paciente decidió continuar con el embarazo.
A las 37.4 semanas se atendió el parto, en un centro de tercer nivel de atención, con recién nacido, de peso adecuado para la edad gestacional, buena adaptación neonatal, sin signos de dificultad respiratoria, pero con cianosis generalizada, además de facies dismórfica. El recién nacido permaneció hospitalizado en la unidad de cuidados intensivos neonatales para tratamiento médico.
Durante su hospitalización fue valorado por el oftalmólogo pediátrico, con hallazgos normales para su edad. Los cardiólogos le practicaron un nuevo ecocardiograma que confirmó los hallazgos, además de una CIA tipo ostium secundum. Los estudios de extensión, para descartar síndromes genéticos asociados y malformaciones extracardiacas, no pudieron practicarse debido a la complejidad de la afección y la necesidad de atención especializada y el requerimiento de un mayor nivel de atención. El recién nacido falleció, sin autopsia. Se refirió, como causa de muerte, el diagnóstico de sepsis.
DISCUSIÓN
Como está descrito en la bibliografía, la prevalencia de malformaciones mayores a nivel internacional corresponde a un 2 a 4%, porcentaje que varía dependiendo de la edad de la población evaluada y si el diagnóstico se estableció antes del nacimiento o luego de ésta,8 lo que supone un grave problema de salud y social. Es indudable que la correcta evaluación ecográfica prenatal permite detectar hasta el 90% de las enfermedades cardiacas graves, cuando la practican especialistas experimentados.9
Una vez sospechado el diagnóstico es necesario descartar causas cromosómicas-genéticas para que el proceso de atención médica sea integral, dirigido, que permita establecer opciones terapéuticas tempranas, con seguimiento y pronóstico precisos y, sobre todo, la prevención de la aparición de nuevos casos mediante un adecuado consejo genético familiar.10
Históricamente, la técnica más utilizada para el análisis cromosómico ha sido el cariotipo, que detecta grandes pérdidas o ganancias de material genético y reordenamientos estructurales hasta en un 5% de los pacientes. Sin embargo, para mejorar el rendimiento diagnóstico se han implementado técnicas, como los arrays de hibridación genómica comparativa, que permiten explorar múltiples loci del genoma de manera simultánea y compararlos con la población sana.10 Esos ensayos de hibridación de microarrays son la herramienta fundamental para la detección de deleciones y duplicaciones.
Los estudios de deleciones y duplicaciones sugieren que los efectos fenotípicos de los cambios en la cantidad de copias son pleiotrópicos e implican la existencia de vías biológicas compartidas entre múltiples condiciones del neurodesarrollo, lo que puede explicar la variabilidad de las manifestaciones que pueden manifestar los pacientes.11
Esta herramienta tiene ventajas frente a las convencionales porque permite una mayor resolución en la detección de alteraciones o cambios en la cantidad de copias, permitie localizar con precisión la alteración, su tamaño y el contenido del gen, lo que facilita un diagnóstico genético más efectivo, preciso y mayor predicción de la enfermedad asociada.12
Teniendo en cuenta lo anterior, y en virtud de la necesidad de evaluar las variantes genéticas encontradas en diferentes enfermedades, así como la interacción de los genes entre sí, surge la necesidad de incluir a la bioinformática como el camino que aporte pistas de mecanismos moleculares, vías de interacción, arquitectura molecular y modelos de predicción de enfermedades, además de ayudar a descubrir nuevas dianas terapéuticas y fármacos.13
La bioinformática es una disciplina científica, emergente, en las ciencias biomédicas que utiliza la tecnología de la información para organizar, analizar y distribuir información biológica, utilizando ADN, ARN, secuencias de aminoácidos, proteínas, estructuras moleculares tridimensionales, interacciones de genes, vías metabólicas, entre otros. Surgió a partir de las bases de datos obtenidas del Proyecto Genoma Humano y a través de la experimentación in silico, permite comprender mejor la relación entre la herencia y el riesgo de padecer una enfermedad, revelando la influencia genética en la aparición y evolución de enfermedades, así como desarrollar pruebas diagnósticas y alternativas terapéuticas para estas afecciones.14 Además, reconoce los genes que se encuentran en regiones eliminadas o insertadas, se realizan análisis en bases de datos GeneMania y/o STITCH en la búsqueda de anotaciones funcionales de genes candidatos, características de genes involucrados en enfermedades conocidas, redes de regulación genética e interacciones proteína-proteína, que permitirán establecer una correlación genotipo-fenotipo.
En la microdeleción 1q21.1, encontrada en el paciente del caso, se ha reportado una amplia gama de características clínicas: rasgos faciales dismórficos leves-variables asociados con prominencia frontal, ojos hundidos, pliegues epicánticos, puente nasal grande, filtrum largo, paladar muy arqueado y trigonocefalia (mayor del 75%); retraso del desarrollo leve a moderado, incluido el retraso motor y del habla (50 a 75%); microcefalia, baja estatura, discapacidad intelectual y anomalías oculares de microftalmia, colobomas coriorretinianos y del iris, estrabismo y cataratas (25-50%); trastorno por déficit de atención con hiperactividad (TDAH), enfermedad congénita por calor, hipotonía, retraso del crecimiento y convulsiones (10 - 25%); y trastornos del espectro autista (TEA), esquizofrenia, malformaciones cerebrales, anomalías esqueléticas, malformaciones genitourinarias y sordera neurosensorial en menos del 10% de los pacientes.11
La cardiopatía congénita (CHD) es una característica importante de la microdeleción 1q21.1 y, ocasionalmente, también se ha informado de su existencia en la duplicación 1q21.1. Según Digilio y colaboradores se ha informado que los individuos con la microdeleción recurrente 1q21.1 tienen cardiopatías; entre ellas: conducto arterioso persistente, tronco arterioso, defectos del tabique auricular y ventricular, tetralogía de Fallot, válvula aórtica bicúspide, dilatación de la aorta ascendente, insuficiencia aórtica, coartación de la aorta, interrupción del arco aórtico, origen anómalo de la arteria coronaria derecha, estenosis de la válvula pulmonar y transposición de los grandes vasos en individuos con deleciones. Hace poco se documentó una asociación entre la tetralogía de Fallot y la duplicación 1q21.1 o variantes en el mapeo del gen GJA5.15
La deleción del cromosoma 1q21.1 fue propuesta, por primera vez, como causa de enfermedad coronaria por Christiansen y su grupo, quienes encontraron deleciones que abarcaban toda la región crítica de 1q21.1 en un caso de enfermedad coronaria sindrómica y dos no sindrómicas entre 505 pacientes derivados para la evaluación genética clínica por sospecha de síndrome de DiGeorge o Williams con obstrucción del arco aórtico como parte de su fenotipo.16
Christiansen y coautores evaluaron 505 casos de pacientes con defectos congénitos a los que se les hizo un estudio citogenético que encontró una microdeleción en el cromosoma 1q21.1 asociada con la expresión génica GJA5, GJA8, FMO5, CHD1L, BCL9, ACP6.16 Entre las alteraciones cardiacas encontradas en este estudio están: defectos del tabique ventricular, estenosis subaórtica, coartación de la aorta.16 Al compararlo con el caso aquí reportado, se evidencian los mismos genes involucrados, lo que puede explicar la amplia heterogeneidad en la presentación clínica.
Digilio y su grupo reportaron una serie de pacientes con alteraciones cardiacas, donde la más común fue la estenosis aórtica, además de talla baja, manifestaciones neurológicas: convulsiones y retraso mental. En el análisis citogenético se encontró una duplicación en el cromosoma 1q21.1, con genes asociados GJA5, CHD1L, PRKAB2. Así mismo, al comparar con el caso que aquí se reporta puede observarse la amplia heterogeneidad en cuanto al espectro de presentación de afección cardiaca asociada con las variantes en los genes.15
Soemedi y colaboradores evaluaron las características fenotípicas específicas asociadas con las variantes en el cromosoma 1q21.1 que involucraban la sobreexpresión del gen GJA5. Ellos encontraron que, en términos de fenotipos de enfermedad cardiaca, los más comunes fueron: transposición de grandes arterias, defectos del tabique auricular, displasia de la válvula mitral y defectos del tabique ventricular.17
Upadhyai y coautores reportaron el caso de un paciente con dismorfia facial, microcefalia, baja estatura, microretrognatia, estrabismo, orejas grandes, labio y paladar hendido, microftalmia, microcórnea. Al hacer el análisis encontraron una deleción en el cromosoma 1q21.1 con los siguientes genes involucrados: NBPF11, PRKAB2, FMO5, CHD1L, BCL9, ACP6, GJA5, GJA8.18 A pesar de tener la misma variante genética que el paciente del caso aquí comunicado, en ese paciente no se detectó cardiopatía congénita, lo que sigue apuntando a la hipótesis de una amplia heterogeneidad en cuanto a la presentación fenotípica de las diferentes interacciones genéticas.
CONCLUSIONES
La microdeleción 1q21.1 es una alteración cromosómica rara en la que se ven afectados múltiples genes, lo que puede traducirse en una gran variedad de alteraciones fenotípicas. Existe, además, un amplio rango de presentación en cuanto a afectación cardiaca y su asociación con otros síndromes genéticos, lo que deja de manifiesto la importancia de los estudios genéticos como método de elección para su identificación y correcta atención multidisciplinaria, además de poder resaltar la importancia de la medicina 5P (personalizada, predictiva, preventiva, participativa y poblacional).
Con base en los hallazgos mencionados es relevante hacer hincapié en la importancia de establecer correctamente un diagnóstico preconcepcional, tanto con las herramientas básicas disponibles durante el control prenatal como la ecografía, así como la evaluación genética necesaria, como la implementación de arrays como método de evaluación de interacción genética que, finalmente, permita tener un diagnóstico claro, para así generar estrategias de tratamiento postnatal, además de una asesoría preconcepcional multidisciplinaria para futuros embarazos.
REFERENCIAS
- Domínguez-Manzano P, Mendoza A, Herraiz I, Escribano D, et al. Transposition of the great arteries in fetal life: diagnostic accuracy and short-term outcome.. Fetal Diagn Ther 2016; 40 (4): 268-76. https://doi.org/10.1159/000444296
- Freire G, Miller M, Huhta J. Ecocardiografía fetal de la transposición de las grandes arterias y el tronco arterial común. Cardiol Young 2012; 22 (6): 671-6. https://doi.org/ 10.1017/S104795111200162X
- C AAR. Una guía práctica para la ecocardiografía fetal: corazones normales y anormales. Transposición de las grandes arterias. 5-Minute Pediatr Consult 8.ª edición, 2022; 4ª ed. (capítulo 37): 956-7.
- Lee MY, Won HS, Han YJ, Ryu HM, et al. Clinical value of chromosomal microarray analysis in prenatally diagnosed dextrotransposition of the great arteries. J Matern Neonatal Med 2020; 33 (9): 1480-5. https://doi.org/ 0.1080/14767058.2018.1519800.
- Edwards SD, Schulze KV, Rosenfeld JA, Westerfield LE, et al. Clinical characterization of individuals with the distal 1q21.1 microdeletion. Am J Med Genet Part A. 2021; 185 (5): 1388-98. https://doi.org/10.1002/ajmg.a.62104
- Wilczewski CM, Obasohan J, Paschall JE, Zhang S, et al. Genotype first: Clinical genómica research through a reverse phenotyping approach. Am J Hum Genet 2023; 110 (1): 3-12. https://doi.org/10.1016/j.ajhg.2022.12.004
- De Ita M, Cisneros B, Rosas-Vargas H. Genética de la transposición de grandes arterias: entre la anormalidad de lateralidad y el defecto del tracto de salida. Revista de Cardiología 2021; 14 (3): 390-9.
- Vargas P, Mergudich T, Martinovic C, Córdova V, et al. Diagnóstico prenatal de malformaciones congénitas y alteraciones cromosómicas: resultado de la experiencia CIMAF, Hospital Dr. Sótero Del Río. Rev Chil Obstet Ginecol 2020; 85 (4): 358-65. http://dx.doi.org/10.4067/S0717-75262020000400358
- Muñoz H, Copado Y, Díaz C, Muñoz G, et al. Diagnóstico prenatal y manejo de la enfermedad cardíaca fetal. Rev Médica Clínica Las Condes 2016; 27 (4): 447-75. http://dx.doi.org/10.1016/j.rmclc.2016.07.006
- Castells-Sarret N, Cueto-González AM, Borregan M, López-Grondona F, et al. Array CGH como primera opción en el diagnóstico genético: 1.000 casos y análisis de coste-beneficio. Una Pediatría 2018; 89 (1): 3-11. https://doi.org/10.1016/j.anpedi.2017.07.011
- Bernier R, Steinman KJ, Reilly B, Wallace AS, et al. Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet Med 2016; 18: 341-349. https://doi.org/10.1038/gim.2015.78
- Querejeta ME, Nieva B, Navajas J, et al. Diagnóstico prenatal y array-CGH II: gestaciones de bajo riesgo. Diagnóstico Prenatal 2012; 23 (2): 49-55. https://doi.org/10.1016/j.diapre.2012.01.004
- Persidis A. Data miniming in biotechnology. Nat Biotechnol 2000; 18 (2): 237-8. https://doi.org/10.1038/72722
- Oliver GR, Hart SN, Klee EW. Bioinformatics for clinical next generation sequencing. Clin Chem 2015; 61 (1): 124-35. https://doi.org/10.1373/clinchem.2014.224360
- Digilio MC, Bernardini L, Consoli F, Lepri FR, et al.Congenital heart defects in recurrent reciprocal 1q21.1 deletion and duplication syndromes: rare association with pulmonary valve stenosis. Eur J Med Genet 2013; 56 (3): 144-9. https://doi.org/10.1016/j.ejmg.2012.12.004
- Christiansen J, Dyck JD, Elyas BG, et al. Chromosome 1q21.1 contiguous gene deletion is associated with congenital heart disease. Circ Res 2004; 94 (11): 1429-35. https://doi.org/ 10.1161/01.RES.0000130528.72330.5c.
- Soemedi R, Topf A, Wilson IJ, Darlay R, et al. Phenotype-specific effect of chromosome 1q21.1 rearrangements and GJA5 duplications in 2436 congenital heart disease patients and 6760 controls. Hum Mol Genet 2012; 21 (7): 1513-20. https://doi.org/
- Upadhyai P, Amiri EF, Guleria VS, et al. Recurrent 1q21.1 deletion syndrome: report on variable expression, nonpenetrance and review of literature. Clin Dysmorphol 2020; 29 (3): 127-31. https://doi.org/10.1097/MCD.0000000000000327


